Conformations of a cyclohexane¶
The geometry optimsation procedures in QM software involve local optimisation of the energy, not global optimisation. This allows us to investigate energy differences between different conformations.
We are going to look at the difference in energy between conformations of 1,4-dichloro-cyclohexane.
Create an initial structure¶
Under Build/Insert/Fragment, choose share/avogadro/fragments/cyclic alkanes/cyclohexane.cml. Click Insert Fragment. Choose Edit/Select None to clear the selection.
Note
Which conformation of cyclohexane is this?
Choose the Draw Tool ( ), and change any hydrogen to a chlorine. Find the hydrogen atom on the opposite carbon that is on the same face of the ring, and change this also to chlorine.
), and change any hydrogen to a chlorine. Find the hydrogen atom on the opposite carbon that is on the same face of the ring, and change this also to chlorine.
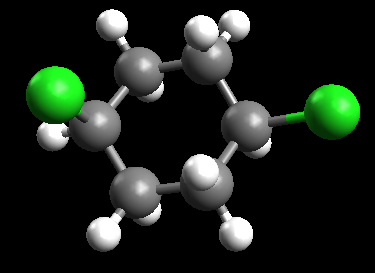
Optimize the geometry using MMFF94.
Note
What parts of the structure change?
Generate conformations interactively¶
Click on the Auto Optimization Tool ( ) and choose Start to turn it on. (Make sure that it is using the MMFF94 forcefield.)
) and choose Start to turn it on. (Make sure that it is using the MMFF94 forcefield.)
Click on one of the chlorine atoms and drag it to change to another conformation.
Note
How many conformations of this cyclohexane can you find?
For each conformation:
- use the Measure Tool (
 ) to measure the chlorine-chlorine distance
) to measure the chlorine-chlorine distance - use Extensions/Molecular Mechanics/Calculate Energy to find the MMFF94 energy
- save each conformation to a CML file
Turn off the Auto Optimization by clicking Stop.
Calculate accurate energies¶
Open the saved conformations one-by-one, and generate input files for GAMESS for a geometry optimisation at the HF/3-21G level of theory.
Carry out the calculation for each conformation and note the energy.
Note
Is the same trend in energies observed for MMFF94 and HF/3-21G?